Forschung
Unsere Forschung befasst sich mit Analysen zur molekularen Pathogenese verschiedener, seltener genetischer Erkrankungen und untersucht die physiologischen Funktionen kurzer Peptide.
Sphingolipide, Fettsäure-2-Hydroxylase und Spastische Paraplegie
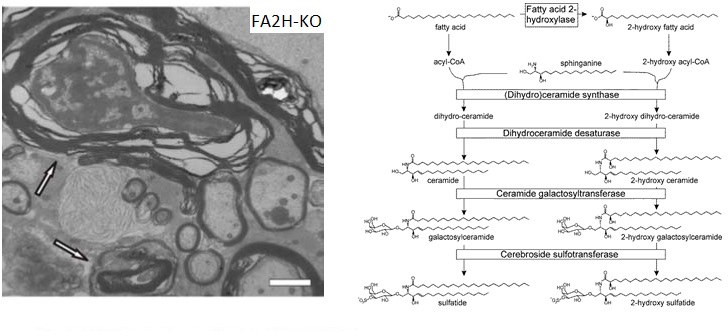
Das Enzym Fettsäure-2-Hydroxylase (FA2H) ist essenziell für die Synthese einer kleinen, aber in einigen Geweben (besonders im Nervensystem und dort im Wesentlichen in den Myelin-produzierenden Zellen, den Oligodendrozyten und Schwann Zellen) häufigen Gruppe von hydroxylierten Sphingolipiden (Sphingolipide sind wichtige Membranlipide in allen Zellen). Defekte im FA2H-Gen sind Ursache für eine Form der Spastischen Paraplegie (SPG35; auch bekannt als FAHN [fatty acid hydroxylase-associated neurodegeneration]). Wir konnten FA2H-defiziente Mäuse als Tiermodell dieser Erkrankung etablieren [Zöller et al., 2008, The Journal of Neuroscience 28:9741-9754] und untersuchen mithilfe dieser Tiere die molekularen Mechanismen der Erkrankung [Hardt et al., 2020, Hum. Mol. Genet. 29:3616-3630; Jordans et al., 2022, Mol. Neurobiol. 59:3969-3979]. Außerdem werden In-vitro-Studien an Zelllinien durchgeführt, um Hypothesen zur Funktion von hydroxylierten Sphingolipiden und zur Pathogenese der FA2H-Defizienz zu testen.
N-Acetylaspartat (NAA) und Morbus Canavan
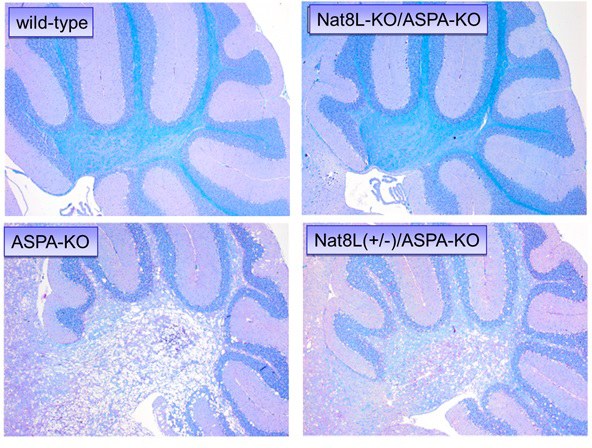
Morbus Canavan ist eine seltene genetische Erkrankung des zentralen Nervensystems, bei der es zu einem zunehmenden Verlust an der weißen Substanz kommt (Leukodystrophie). Charakteristischerweise kommt es bei der Erkrankung zu einer schwammartigen Degeneration im Gehirn. Die Ursache der Erkrankung ist ein Defekt in dem Gen ASPA; dieses Gen kodiert für das Enzym Aspartoacylase, welches die natürlicherweise im Gehrin vorkommende Verbindung N-Acetylaspartat (NAA) abbaut. Fehlt das Enzym, akkumuliert das NAA in immer höheren Konzentrationen im Gehirn. Mit unseren Forschungen an einem Mausmodell der Erkrankung (Aspartoacylase-defiziente Mäuse, ASPA-KO) konnten wir zeigen, dass ein zusätzlicher Mangel an dem Gen Nat8L, das für die Synthese von NAA verantwortlich ist, die schwammartige Degeneration verhindert [Maier et al., 2015, The Journal of Neuroscience 35:14501-14516]. Damit konnten andere Hypothesen zur Pathologie der Erkrankung widerlegt werden und ein Weg für eine neue therapeutische Option (Hemmung des Nat8L-Enzyms) aufgezeigt werden.
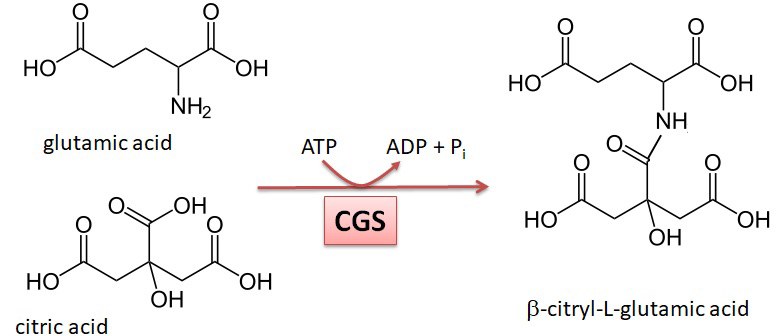
Citrylglutamat und Spermatogenese
β-Citrylglutamat ist ein kleines Pseudopeptid, das in vielen Geweben von Wirbeltieren vorkommt, in besonders hohen Konzentrationen aber im Hoden. Wir konnten zeigen, dass β-Citrylglutamat essenziell für eine normale Bildung von Spermien ist; männliche Mäuse, die nicht mehr in der Lage sind, β-Citrylglutamat zu synthetisieren, sind subfertil bis infertil [Wang-Eckhardt et al., 2022, Biochemical Journal 479:853-972]. Weitere Untersuchungen zeigten, dass ein später Schritt der Spermiogenese, bei dem die Histone des Chromatins durch andere Proteine (Transition-Proteine, Protamine) ersetzt werden, um eine dichtere Verpackung des Erbmaterials zu ermöglichen, in Abwesenheit von β-Citrylglutamat defekt ist. Mit unseren Arbeiten versuchen wir, die Funktion dieses Pseudopeptids auf molekularer Ebene zu verstehen.