Forschung
Metachromatische Leukodystrophie (MLD) ist eine lysosomale Speichererkrankung, die durch eine funktionelle Defizienz der Arylsulfatase A (ASA) ausgelöst wird. ASA katalysiert den Abbau von Sulfatid, einem Sphingolipid, das vor allem in Myelinscheiden vorkommt. Fehlt ASA, akkumuliert Sulfatid in Oligodendrozyten und Schwann-Zellen. Dadurch kommt es zu einer fortscheitenden Demyelinisierung des peripheren und zentralen Nervensystems. Bei der typischen Verlaufsform zeigen MLD-Patienten mit etwa 2 Jahren erste klinische Symptome, wie Gangunsicherheiten, Ataxie, Sprachverlust und Lähmungen. Die sich verschlimmernde zentralnervöse Symptomatik führt vor dem 9. Lebensjahr zum Tode.
Therapie der MLD
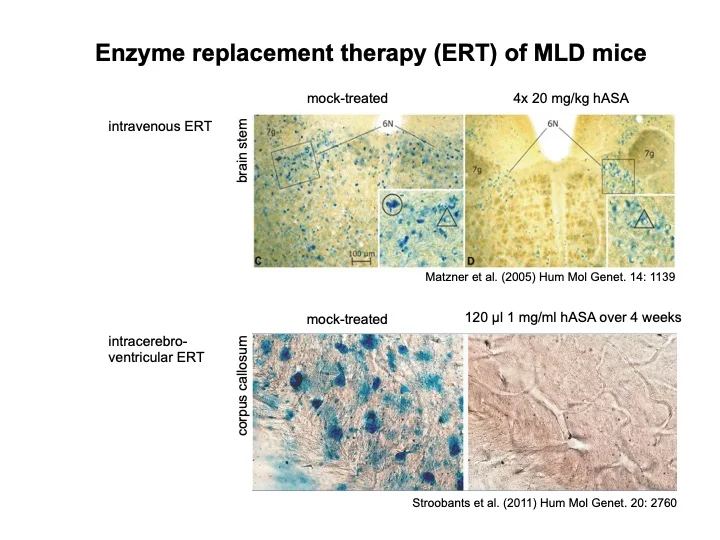
Wir arbeiten an einer Therapie der MLD. Die N-Glykane der ASA tragen Mannose 6-phosphat (M6P) Reste, über die das Enzym von M6P-Rezeptoren an der Plasmamembran gebunden und zum Lysosom transportiert wird. Die M6P-abhängige Endozytose eröffnet die Möglichkeit, MLD durch Enzymsubstitutionstherapie oder Gentherapie zu behandeln. Bei der Enzymersatztherapie wird rekombinant hergestellte, enzymatisch aktive ASA injiziert. Anhand eines von uns entwickelten ASA knockout Maus-Modells der MLD haben wir gezeigt, dass Infusion von ASA entweder in die Blutbahn oder in die Cerebrospinalflüssigkeit zu einer Abnahme der Sulfatidspeicherung im Zentralnervensystem führt. Unsere präklinischen Untersuchungen führten zu mehreren klinischen PhaseI/II Studien, die derzeit in den USA und Europa durchgeführt werden.
Steigerung des therapeutischen Potenzials der Enzymersatztherapie und Gentherapie
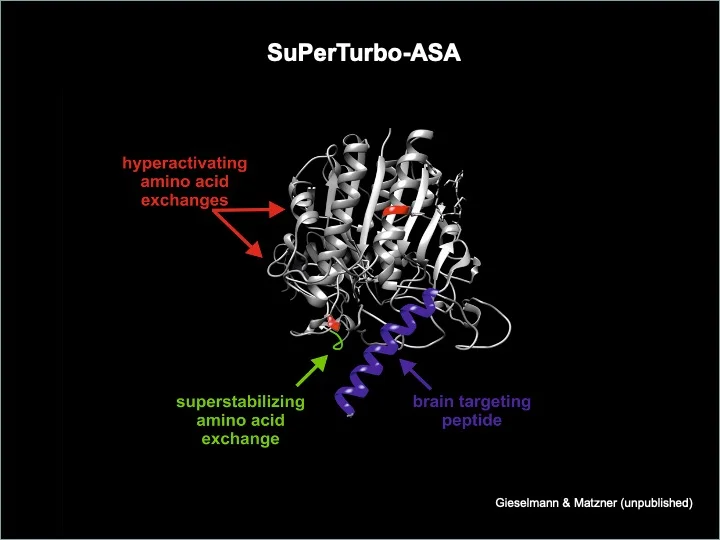
Durch Genetic Engineering der ASA streben wir eine Steigerung des therapeutischen Potenzials der Enzymersatztherapie und Gentherapie an. Zur Verbesserung des Übertritts von ASA aus der Blutbahn in das Gehirnparenchym haben wir ein Peptid entwickelt, das die ASA aktiv über die Bluthirnschranke transportiert. Fusionsproteine zwischen ASA und diesem Peptid führen im Tiermodell zu einer annähernden Verdopplung der therapeutischen Effizienz. Weiterhin konnten wir durch gezielte Aminosäureaustäusche innerhalb der ASA-Domäne sowohl die katalytische Rate als auch die extra- und intrazelluläre Halbwertszeit des Enzyms erheblich steigern. Die resultierende trimodal optimierte ASA-Variante wurde von uns unter der Bezeichnung SuPerTurbo-ASA patentiert und soll in Kooperation mit europäischen und US-amerikanischen Pharmaunternehmen klinisch getestet werden.
Pathomechanismus der MLD
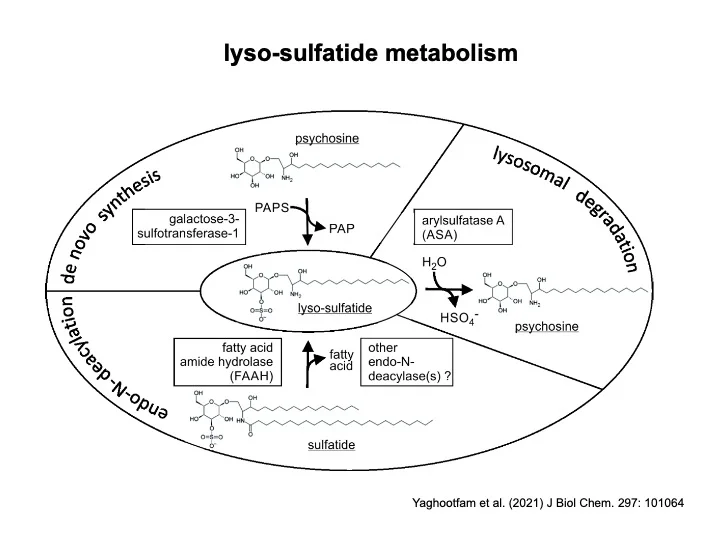
Mit dem Ziel bisher unerkannte therapeutische Targets zu identifizieren, beschäftigen wir uns in einem weiteren Forschungsschwerpunkt mit dem Pathomechanismus der MLD. Nach unserer Arbeitshypothese lösen neuroinflammatorische Prozesse den Verlust der Myelinscheide aus. Tatsächlich verzögert eine Behandlung von MLD-Mäusen mit antiinflammatorischen Wirkstoffen die Demyelinisierung. Antiphlogistika könnten damit ergänzend zur Enzymersatztherapie oder Gentherapie eingesetzt werden. Die Neuroinflammation wird wahrscheinlich nicht durch Sulfatid selbst, sondern durch einen als lyso-Sulfatid bezeichneten Metaboliten ausgelöst. Die Stoffwechselwege, die zur Bildung des cytotoxischen lyso-Sulfatids führen, werden von uns untersucht. Inhibitoren, die die lyso-Sulfatid Bildung vermindern, könnten eine weitere Möglichkeit zur Behandlung der MLD darstellen.